Bennett acceptance ratio
The Bennett acceptance ratio method (sometimes abbreviated to BAR) is an algorithm for estimating the difference in free energy between two systems (usually the systems will be simulated on the computer). It was suggested by Charles H. Bennett in 1976.[1]
Contents |
Preliminaries
Take a system in a certain super state. By performing a Metropolis Monte Carlo walk it is possible to sample the landscape of states that the system moves between, using the equation
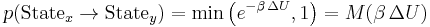
where ΔU = U(Statey) − U(Statex) is the difference in potential energy, β = 1/kT (T is the temperature in Kelvins while k is the Boltzmann constant), and 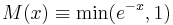 is the Metropolis function. The resulting states are then sampled according to the Boltzmann distribution of the super state at temperatue T. Alternatively, if the system is dynamically simulated in the canonical ensemble (also called the NVT ensemble), the resulting states along the simulated trajectory are likewise distributed. Averaging along the trajectory (in either formulation) is denoted by angle brackets
is the Metropolis function. The resulting states are then sampled according to the Boltzmann distribution of the super state at temperatue T. Alternatively, if the system is dynamically simulated in the canonical ensemble (also called the NVT ensemble), the resulting states along the simulated trajectory are likewise distributed. Averaging along the trajectory (in either formulation) is denoted by angle brackets 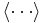 .
.
Suppose that two super states of interest, A and B, are given. We assume that they have a common configuration space, i.e., they share all of their micro states, but the energies associated to these (and hence the probabilities) differ because of a change in some parameter (such as the strength of a certain interaction). The basic question to be addressed is, then, how can the Helmholtz free energy change (ΔF = FB − FA) on moving between the two super states be calculated from sampling in both ensembles? Note that the kinetic energy part in the free energy is equal between states so can be ignored. Note also that the Gibbs free energy corresponds to the NpT ensemble.
The general case
Bennett shows that for every function f satisfying the condition 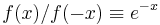 (which is essentially the detailed balance condition), and for every energy offset C, one has the exact relationship
(which is essentially the detailed balance condition), and for every energy offset C, one has the exact relationship
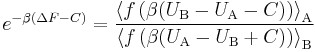
where UA and UB are the potential energies of the same configurations, calculated using potential function A (when the system is in superstate A) and potential function B (when the system is in the superstate B) respectively.
The basic case
Substituting for f the Metropolis function defined above (which satisfies the detailed balance condition), and setting C to zero, gives
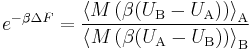
The advantage of this formulation (apart from its simplicity) is that it can be computed without performing two simulations, one in each specific ensemble. Indeed, it is possible to define an extra kind of "potential switching" Metropolis trial move (taken every fixed number of steps), such that the single sampling from the "mixed" ensemble suffices for the computation.
The most efficient case
Bennett explores which specific expression for ΔF is the most efficient, in the sense of yielding the smallest standard error for a given simulation time. He shows that the optimal choice is to take
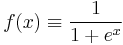 , which is essentially the Fermi–Dirac distribution (satisfying indeed the detailed balance condition).
, which is essentially the Fermi–Dirac distribution (satisfying indeed the detailed balance condition).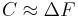 . This value, of course, is not known (it is exactly what we are trying to compute), but it can be approximately chosen in a self consistent manner.
. This value, of course, is not known (it is exactly what we are trying to compute), but it can be approximately chosen in a self consistent manner.
Some assumptions needed for the efficiency are the following:
- The densities of the two super states (in their common configuration space) should have a large overlap. Otherwise, a chain of super states between A and B may be needed, such that the overlap of each two consecutive super states is adequate.
- The sample size should be large. In particular, as successive states are correlated, the simulation time should be much larger than the correlation time.
- The cost of simulating both ensembles should be approximately equal - and then, in fact, the system is sampled roughly equally in both super states. Otherwise, the optimal expression for C is modified, and the sampling should devote equal times (rather than equal number of time steps) to the two ensembles.
Relation to other methods
The perturbation theory method
This method, also called Free energy perturbation (or FEP), involves sampling from state A only. Unsurprisingly, it might be much less efficient than the BAR method. In fact, it requires that all the high probability configurations of super state B are contained in high probability configurations of super state A, which is a much more stringent requirement than the overlap condition stated above.
The exact (infinite order) result
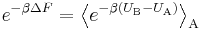
or
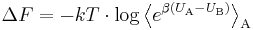
This exact result can be obtained from the general BAR method, using (for example) the Metropolis function, in the limit 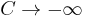 . Indeed, in that case, the denominator of the general case expression above tends to 1, while the numerator tends to
. Indeed, in that case, the denominator of the general case expression above tends to 1, while the numerator tends to 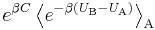 . A direct derivation from the definitions is more straightforward, though.
. A direct derivation from the definitions is more straightforward, though.
The second order (approximate) result
Assuming that 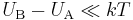 and Taylor expanding the second exact perturbation theory expression to the second order, one gets the approximation
and Taylor expanding the second exact perturbation theory expression to the second order, one gets the approximation
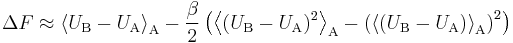
Note that the first term is the expected value of the energy difference, while the second is essentially its variance.
The first order inequalities
Using the convexity of the log function appearing in the exact perturbation analysis result, together with Jensen's inequality, gives an inequality in the linear level; combined with the analogous result for the B ensemble one gets the following version of the Gibbs-Bogoliubov inequality:
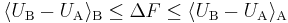
Note that the inequality agrees with the negative sign of the coefficient of the (positive) variance term in the second order result.
The thermodynamic integration method
writing the potential energy as depending on a continuous parameter, 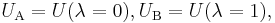
one has the exact result 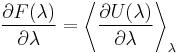 This can either be directly verified from definitions or seen from the limit of the above Gibbs-Bogoliubov inequalities when
This can either be directly verified from definitions or seen from the limit of the above Gibbs-Bogoliubov inequalities when 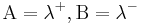 . we can therefore write
. we can therefore write
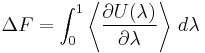
which is the thermodynamic integration (or TI) result. It can be approximated by dividing the range between states A and B into many values of λ at which the expectation value is estimated, and performing numerical integration.
Implementation
The Bennett acceptance ratio method is implemented in modern molecular dynamics systems, such as Gromacs.